Module 2: Gene Expression Validation
A DNA mutation means nothing if it is never translated into protein. This module takes the somatic mutations discovered in Module 1 and cross-references them against RNA-seq data from the same tumor. Only mutations that are actively transcribed, and therefore capable of producing abnormal proteins visible to the immune system, survive this filter.
Prepare RNA-seq Reference
RNA-seq alignment requires a different kind of index than DNA alignment. While DNA reads map continuously along the genome, RNA reads can span splice junctions, the boundaries between exons where introns have been removed during mRNA processing. A standard DNA aligner like BWA would fail to map these junction-spanning reads correctly.
We start by downloading a gene annotation file (GTF) from Ensembl[1] release 104. This file describes the precise genomic coordinates of every known gene, transcript, exon, and coding sequence in the dog genome. STAR[2] uses this information to build a splice-aware genome index that can handle reads crossing exon-exon boundaries.
We also build an RSEM reference[3], which maps gene/transcript IDs from the GTF to the genome sequence. This will be used in Step 05 to quantify expression at both gene and isoform levels.
# Download gene annotation (GTF) from Ensembl release 104
wget $GTF_URL
gunzip Canis_lupus_familiaris.CanFam3.1.104.gtf.gz
mv Canis_lupus_familiaris.CanFam3.1.104.gtf $GTF
# Build STAR genome index
STAR \
--runMode genomeGenerate \
--runThreadN $THREADS \
--genomeDir $STAR_IDX_DIR \
--genomeFastaFiles $REF \
--sjdbGTFfile $GTF_FILE \
--sjdbOverhang 100
# Build RSEM reference
rsem-prepare-reference \
--gtf $GTF_FILE \
$REF \
$RSEM_REF_PREFIXDownload RNA-seq Data
In addition to the DNA sequencing data used in Module 1, we need RNA sequencing (RNA-seq) data from the same tumor. While DNA tells us what mutations exist, RNA tells us which genes are actually being read and converted into proteins. A mutation in a gene that the tumor has silenced will never produce a targetable protein.
The tumor RNA-seq data is downloaded from SRA using fasterq-dump, the same tool used in Module 1. The output is a pair of FASTQ files (R1 and R2) representing the forward and reverse reads of each RNA fragment.
# Download RNA-seq FASTQ from SRA
fasterq-dump --split-files --threads $THREADS $SRR_TUMOR_RNAQC & Trimming
Before alignment, the raw RNA-seq reads go through the same quality control as Module 1. We use fastp[5] to remove adapter sequences, trim bases below Phred quality 20, and discard reads shorter than 36 bp. This ensures only high-quality reads enter the alignment step.
Interactive HTML reports are generated for each sample, providing a visual summary of read quality, adapter content, and filtering results.
# Trim adapters and low-quality bases
fastp \
-i raw/${SRR}_1.fastq -I raw/${SRR}_2.fastq \
-o trimmed/${SRR}_1.trimmed.fastq.gz -O trimmed/${SRR}_2.trimmed.fastq.gz \
--thread $THREADS \
--qualified_quality_phred 20 \
--length_required 36 \
--detect_adapter_for_pe \
--json qc/${SRR}_fastp.json --html qc/${SRR}_fastp.htmlSTAR Alignment
STAR[2] is the gold-standard RNA-seq aligner. Unlike BWA (used for DNA), STAR is designed to handle splice junctions. It can correctly map reads that span the boundary between two exons separated by thousands of bases of intronic sequence.
We use two-pass alignment (--twopassMode Basic). In the first pass, STAR discovers novel splice junctions not present in the annotation. In the second pass, it re-aligns all reads using both annotated and newly discovered junctions, significantly improving sensitivity for novel transcripts.
The --quantMode TranscriptomeSAM GeneCounts flag tells STAR to produce two outputs simultaneously: a genome-aligned BAM (for variant validation in Step 06) and a transcriptome-aligned BAM (for expression quantification in Step 05). This avoids the need for separate alignment runs.
# Align with STAR (two-pass mode)
STAR \
--runThreadN $THREADS \
--genomeDir $STAR_IDX \
--readFilesIn trimmed/${SRR}_1.trimmed.fastq.gz trimmed/${SRR}_2.trimmed.fastq.gz \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--outSAMattrRGline ID:${SRR} SM:${SRR} PL:ILLUMINA \
--outFileNamePrefix alignment/${SRR}_ \
--twopassMode Basic \
--quantMode TranscriptomeSAM GeneCounts
# Index and collect stats
samtools index alignment/${SRR}_Aligned.sortedByCoord.out.bam
samtools flagstat alignment/${SRR}_Aligned.sortedByCoord.out.bamExpression Quantification
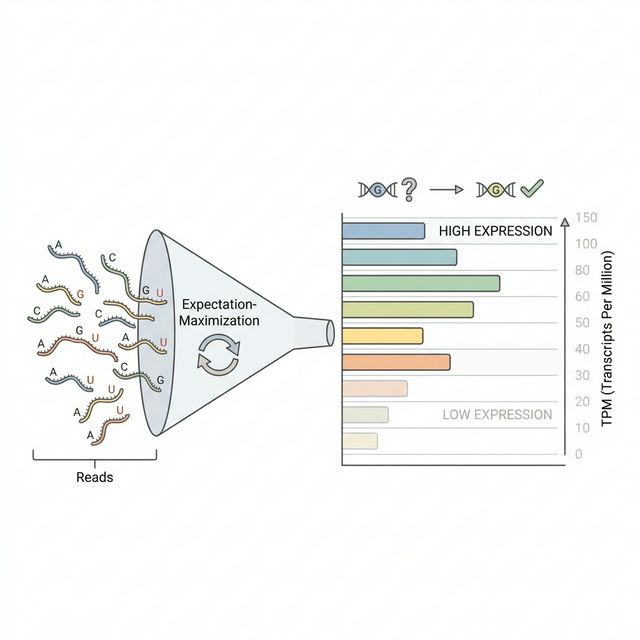
Now that reads are aligned, we need to count how many reads map to each gene. This tells us how actively each gene is being transcribed in the tumor. Genes with high read counts are highly expressed and more likely to produce enough abnormal protein to trigger an immune response.
We use RSEM[3](RNA-Seq by Expectation-Maximization), which takes the transcriptome-aligned BAM from Step 04 and quantifies expression at both gene and isoform levels. RSEM's key strength is its ability to handle multi-mapping reads, reads that could originate from multiple similar genes or transcript variants. Using an expectation-maximization algorithm, RSEM probabilistically distributes these ambiguous reads to their most likely source.
The pipeline automatically detects the library strandednessfrom STAR's ReadsPerGene output. By comparing forward-strand and reverse-strand read counts, it determines whether the library is unstranded, forward-stranded, or reverse-stranded, and passes the correct parameter to RSEM.
The output includes three expression metrics for each gene: expected count (raw read count after EM redistribution), TPM (Transcripts Per Million, normalized for gene length and sequencing depth), and FPKM (Fragments Per Kilobase of transcript per Million mapped reads). TPM is generally preferred for cross-sample comparisons because its values sum to the same total across samples.
# Auto-detect strandedness from STAR ReadsPerGene output
# If reverse >> forward (2x): reverse-stranded
# If forward >> reverse (2x): forward-stranded
# Otherwise: unstranded
STRAND_COUNTS=$(awk 'NR>4 {f+=$3; r+=$4} END {printf "%d %d", f, r}' \
alignment/${SRR}_ReadsPerGene.out.tab)
# Run RSEM on tumor transcriptome BAM
rsem-calculate-expression \
--paired-end \
--alignments \
--no-bam-output \
-p $THREADS \
--strandedness $STRANDEDNESS \
alignment/${SRR}_Aligned.toTranscriptome.out.bam \
$RSEM_REF \
expression/${SRR}Somatic Variant Validation
This is the critical integration step where DNA and RNA evidence converge. We take every somatic PASS variant from Module 1 and ask: "Is this mutation actually visible in the tumor's RNA?"A mutation that exists in DNA but isn't expressed in RNA cannot produce an abnormal protein, and therefore cannot serve as a vaccine target.
Variants are first separated into SNVs and indels, because they require different validation strategies. For SNVs, we use bam-readcount[4] to count allele-specific reads at each position. For indels, we use bcftools mpileup to re-genotype the RNA BAM at each indel site, since bam-readcount cannot reliably handle insertions and deletions.
Each variant is then classified into one of four categories based on RNA evidence:
- VALIDATED: alt reads ≥ 2 and VAF ≥ 5% (strong RNA support)
- WEAK_SUPPORT: alt reads ≥ 1 but below validation threshold
- NOT_EXPRESSED: site has RNA coverage but no alt reads
- NO_COVERAGE: no RNA reads at this position
Only VALIDATED variants advance to the next modules for neoantigen prediction. This filter is essential because it eliminates mutations in genes that are silenced in the tumor, or in regions not captured by RNA-seq, dramatically reducing false-positive neoantigen candidates.
# Separate SNVs and indels
bcftools view -v snps $SOMATIC_VCF -Oz -o somatic_snvs.vcf.gz
bcftools index -t somatic_snvs.vcf.gz
bcftools view -V snps $SOMATIC_VCF -Oz -o somatic_indels.vcf.gz
bcftools index -t somatic_indels.vcf.gz
# Phase 1: Validate SNVs with bam-readcount
bam-readcount -f $REF -l snv_sites.bed -q 20 -b 20 $RNA_BAM > snv_readcounts.tsv
# Phase 2: Validate indels with bcftools mpileup
bcftools mpileup -f $REF -R somatic_indels.vcf.gz \
-a FORMAT/AD,FORMAT/DP --no-BAQ -q 20 -Q 20 -d 10000 $RNA_BAM | \
bcftools call -m -A --ploidy 2 -Ov -o rna_indel_calls.vcf
# Classify each variant: VALIDATED | WEAK_SUPPORT | NOT_EXPRESSED | NO_COVERAGEExpression Integration
A variant might be validated by RNA-seq reads (Step 06), but if its overall gene expression is extremely low, it is unlikely to produce sufficient peptide for MHC presentation later in the pipeline. This step integrates the variant validation results with the gene-level expression metrics from RSEM.
We extract mapping information (Gene ID, Gene Symbol, and Consequence) from the Module 1 VEP-annotated VCF, and cross-reference it with the RSEM outputs to assign a specific TPM (Transcripts Per Million) value to each validated mutation.
Variants that achieved VALIDATED status in Step 06 and demonstrate a solid expression value (TPM ≥ 1.0) are flagged as strong neoantigen candidates and rigorously prioritized for downstream downstream modeling.
# 1. Build gene expression lookup (Gene_ID -> TPM)
awk 'NR>1{print $1"\t"$6}' $GENE_EXPR > gene_tpm_lookup.tsv
# 2. Extract variant-to-gene mapping from VEP annotation
grep -v "^#" $VCF_ANNOTATED | awk -F'\t' '{
# Parses the CSQ string to extract transcript fields
print chr"\t"pos"\t"ref"\t"alt"\t"gene_id"\t"gene_symbol"\t"consequence
}' > variant_gene_map.tsv
# 3. Join validation results, variant-to-gene maps, and TPM counts
# Outputs: CHROM, POS, REF, ALT, TYPE, RNA_DEPTH, RNA_ALT_COUNT, RNA_VAF, STATUS, GENE_ID, GENE_SYMBOL, CONSEQUENCE, TPM
awk '...' > variants_with_expression.tsvExpected Metrics & QC Checkpoints
Before proceeding to MHC typing and neoantigen prediction, verify these quality metrics from Module 2. Poor RNA-seq quality or low alignment rates will compromise expression quantification and variant validation accuracy.
1. RNA-seq Reads (fastp)
- Passing Filter> 90%
- Q30 Bases> 85%
- Total Reads≥ 30M pairs
RNA-seq typically requires at least 30M read pairs for reliable gene-level quantification.
2. STAR Alignment
- Uniquely Mapped> 70%
- Multi-mapped< 15%
- Unmapped< 10%
High unmapped rates may indicate contamination, adapter issues, or reference mismatch.
3. Variant Validation
- VALIDATEDVariable
(Depends on tumor purity) - Min RNA VAF≥ 5%
- Min Alt Reads≥ 2
Low validation rates may indicate the tumor RNA sample doesn't match the WES tissue source.
References
- Cunningham, F. et al. (2022). Ensembl 2022. Nucleic Acids Research, 50(D1), D988-D995. doi:10.1093/nar/gkab1049 [Ensembl GTF annotation]
- Dobin, A. et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15-21. doi:10.1093/bioinformatics/bts635 [STAR aligner]
- Li, B. & Dewey, C.N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. doi:10.1186/1471-2105-12-323 [RSEM]
- Khanna, A. et al. (2022). bam-readcount: rapid generation of basepair-resolution sequence metrics. Journal of Open Source Software, 7(76), 3722. doi:10.21105/joss.03722 [bam-readcount]
- Chen, S. et al. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884-i890. doi:10.1093/bioinformatics/bty560 [fastp]