Module 4: Neoantigen Prediction & Ranking
The ultimate goal of a cancer vaccine is to activate both CD8+ (cytotoxic) and CD4+ (helper) T cells. This module translates the DNA variants discovered in Module 1 into mutant peptides and uses machine learning to predict their binding affinity to the patient's specific MHC Class I and Class II molecules (genotyped in Module 3).
Prepare Proteome & MHC Sequences
To translate our DNA mutations into protein sequences, we first need the complete reference proteome for the dog (CanFam3.1). We download the Ensembl release 104 proteome, which perfectly matches the GTF and VEP cache used in Module 1.
Additionally, we need the exact amino acid sequences of the dog's MHC (DLA) molecules. While human HLA sequences are built into most prediction tools, canine DLA sequences often require manual supply. By downloading the IPD-MHC[1]protein database, we can provide the exact sequences of the patient's DLA alleles to the prediction engine, allowing it to extrapolate binding preferences even for rare or uncharacterized canine alleles.
# Download Ensembl CanFam3.1 proteome
wget -O reference/proteome/$PROTEOME_FA.gz $PROTEOME_URL
gunzip reference/proteome/$PROTEOME_FA.gz
# Download MHC protein sequences from IPD-MHC
wget -O reference/dla/MHC_prot.fasta \
https://raw.githubusercontent.com/ANHIG/IPDMHC/Latest/MHC_prot.fastaGenerate Class I Peptides
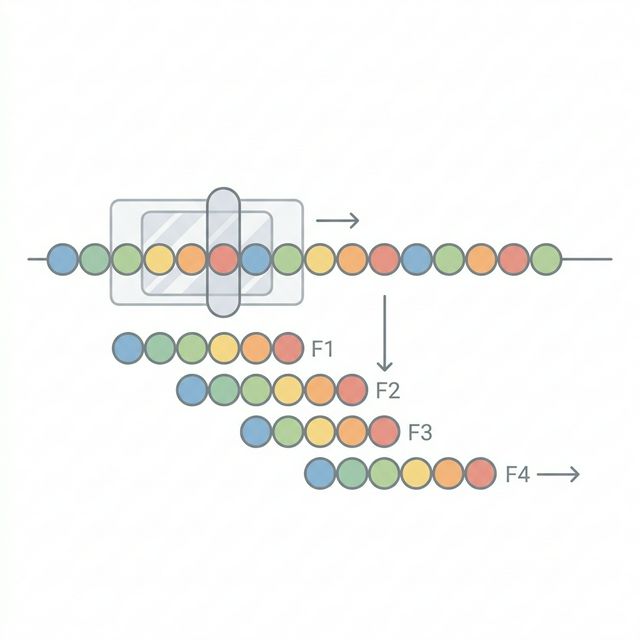
MHC Class I molecules present short peptides of 8 to 11 amino acidsto systemic CD8+ T cells. To identify potential neoantigens, we take the coding variants from Module 1 and simulate the peptides that would be generated by the tumor cell's proteasome.
For each mutation, we extract the surrounding wildtype protein sequence from our reference proteome and apply the amino acid change to create the mutant protein sequence. We then use a sliding window approach to generate every possible 8, 9, 10, and 11-mer peptide that overlaps the mutated position.
This step outputs two lists: the newly formed mutant peptides, and their exact wildtype counterparts. Both are crucial for later prioritization to ensure we target truly "foreign" neo-epitopes.
# 1. Look up variant transcript in reference proteome
# 2. Extract surrounding wildtype sequence & apply mutation
# 3. Generate overlapping 8-11mer peptides
# Example output (sliding window over a mutation):
# WT: SLPQQDIL
# MUT: SLPQQDIV
# WT: LPQQDILK
# MUT: LPQQDIVKClass I Binding Prediction
With our list of mutant peptides and the patient's confirmed DLA Class I alleles (e.g., DLA-88, DLA-12, DLA-64), we use netMHCpan[2] to predict binding affinity.
We utilize the -hlaseqflag, providing the full amino acid sequence of the DLA molecule from IPD-MHC. The network extrapolates the molecule's binding pocket preferences and outputs an Eluted Ligand (EL) score (likelihood of being naturally presented) and a Binding Affinity score (nM). We run predictions for both mutant and wildtype peptide lists.
# Extract sequence for patient's DLA Class I allele
awk -v allele="DLA-88_05101" '...extract sequence...' MHC_prot.fasta > allele.fsa
# Run netMHCpan on mutant and wildtype peptides
netMHCpan -hlaseq allele.fsa -p -f mutant_peptides.pep -BA > mutant_binding.out
netMHCpan -hlaseq allele.fsa -p -f wildtype_peptides.pep -BA > wildtype_binding.outClass I Prioritization
To find the best Class I vaccine candidates, we filter for "strong binders" (EL score ≥ 0.5) and "weak binders" (EL score ≥ 0.1). But binding is not the only metric.
We calculate the Differential Agretopicity Index (DAI)[3], which is the mutant EL score minus the wildtype EL score. A high DAI implies the mutation created a completely new binding motif, making it highly foreign to the immune system. We then merge these predictions with Module 2 RNA-seq findings to ensure the underlying gene is actively transcribed.
# 1. Filter binders (Mutant EL_SCORE >= 0.1)
awk -F'\t' '$5 >= 0.1 && $11=="MUT"' binding_results.tsv > binders.tsv
# 2. Calculate DAI (DAI = Mutant_EL_Score - Wildtype_EL_Score)
# 3. Annotate with RNA validation status
# 4. Sort by mutant EL_SCORE (descending)Generate Class II Peptides
MHC Class II molecules present longer peptides (typically 15-mers) to CD4+ T helper cells. A robust vaccine requires CD4+ help to sustain the CD8+ cytotoxic response.
We repeat the sliding window process from Step 02, but this time generating overlapping 15-mer peptides (using a wider 30 amino acid flank). Just as before, we create paired lists of mutant and wildtype 15-mers mapping to the coding somatic variants.
# Generate 15-mer sliding windows
# Extracted flanking region increased to ±40 amino acids to accommodate longer windows
for ((S = MUT_POS - K + 1; S <= MUT_POS + ALT_AA_LEN - 1; S++)); do
# K=15
MUT_PEP="${MUT_REGION:$((S-1)):15}"
WT_PEP="${WT_REGION:$((S-1)):15}"
doneClass II Binding Prediction
Class II prediction is more complex because the binding groove is open at both ends, and the molecule itself is often a heterodimer composed of an Alpha chain and a Beta chain (e.g., DQA1 and DQB1).
We use netMHCIIpan to predict Class II binding. For single-chain loci (like DRB1, assuming a monomorphic DRA), we run it with a single -hlaseq. For heterodimeric loci, we provide both sequences using -hlaseqA and -hlaseq simultaneously, testing every possible DQA1 + DQB1 pair the patient possesses.
# 1. DRB1 prediction (single-chain approximation)
netMHCIIpan -hlaseq DRB1_allele.fsa -p -f mutant_class2.pep -BA > DRB1_mutant.out
# 2. DQA1 + DQB1 Heterodimeric prediction (Alpha + Beta)
netMHCIIpan -hlaseqA DQA1_allele.fsa -hlaseq DQB1_allele.fsa \
-p -f mutant_class2.pep -BA > DQ_mutant.outClass II Prioritization
Class II binding is inherently weaker and more promiscuous than Class I binding. Therefore, the filtering thresholds are adjusted: a strong binder is still EL ≥ 0.5, but a weak binder is EL 0.1 - 0.5, and we even consider marginal binders (EL 0.05 - 0.1) as valid CD4+ candidates depending on DAI and RNA expression.
# Filter Class II binders (Mutant EL_SCORE >= 0.05)
awk -F'\t' '$5 >= 0.05 && $7=="MUT"' class2_binding_results.tsv > class2_binders.tsv
# Calculate DAI and annotate with RNA validation as done in Class I.Expected Metrics & QC Checkpoints
The quality of neoantigen predictions depends heavily on the previous modules. Review these metrics to ensure the final candidate list is robust:
1. Peptide Generation
- Class I Peptides1,000 - 3,000
- Class II Peptides400 - 1,000
- Windows / Variant~38 (C1), 15 (C2)
A typical missense mutation yields ~38 Class I (8-11mer) and 15 Class II (15-mer) peptides.
2. MHC Binding Success
- Prediction RunCompleted without errors
- Class I Binders (EL≥0.1)~ 2-5%
- Class II Binders (EL≥0.05)~ 5-10%
Check logs if prediction success is 0; IPD-MHC may be missing the DLA allele sequence.
3. Final Candidates
- High DAIDAI > 0
- RNA Validated> 0 candidates
The best candidates are strong mutant binders with NO wildtype binding (High DAI) and active tumor expression.
References
- Maccari, G. et al. (2017). IPD-MHC 2.0: an improved inter-species database for the study of the major histocompatibility complex. Nucleic Acids Research, 45(D1), D860–D864. doi:10.1093/nar/gkw1050 [IPD-MHC]
- Reynisson, B. et al. (2020). NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Research, 48(W1), W449–W454. doi:10.1093/nar/gkaa379 [netMHCpan & netMHCIIpan]
- Duan, F. et al. (2014). Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules for tumor rejection. Journal of Experimental Medicine, 211(11), 2231–2248. doi:10.1084/jem.20141308 [Differential Agretopicity Index / DAI]